基于原位阳离子交换策略的TiO2 光阳极表面Co单原子构筑及其高效稳定太阳能水裂解

https://www.sciencedirect.com/science/article/pii/S0926337323002734
研 究 背 景
光电催化技术(PEC)能够将太阳能转化为高能量密度的氢能,是缓解能源和环境问题的一种极具前景的解决方案。PEC的整体性能往往受到光阳极较差的光收集能力、低的电荷分离和转移效率、以及缓慢的水氧化动力学所限制。因此,优异光阳极的材料的探索成为该领域研究热点。其中,TiO2因其具有合适的能带结构,以及无毒,低成本与耐光腐蚀等优势,而被公认为是最具前景的候选材料之一。然而,TiO2光阳极存在着吸光性差,析氧反应动力学十分缓慢,且存在严重的表面电荷复合。
文 章 简 介
针对上述TiO2光阳极研究领域所存在的科学问题,近日,宁波工程学院侯慧林副教授、杨为佑研究员与天津大学的何芳教授合作,在国际知名期刊“Applied Catalysis B: Environmental”上发表题为:In-situ cation-exchange strategy for engineering single-atomic Co on TiO2 photoanode toward efficient and durable solar water splitting”的研究论文(DOI: 10.1016/j.apcatb.2023.122630)。该研究工作基于一种原位气相阳离子置换材料来调控TiO2光阳极的表面态,实现Co单原子活性位点在TiO2光阳极表面的有效构筑,探究了所构筑光阳极材料在模拟太阳光下的水氧化性能及其机理。基于材料实验表征、水氧化性能的系统测试以及DFT计算等手段,证实了Co单原子活性位点引入可实现光生电子-空穴对的有效分离以及材料可见光波段吸收的提高,同时亦可降低水氧化的反应势垒,所制备的Co/TiO2光阳极的光电流密度达1.47 mA cm-2 (1.23 V vs.RHE) , 是纯TiO2 光阳极3倍,并展示出长达100 h的稳定性。该工作为先进光电阳极材料的理性设计提供了一定的新思路。
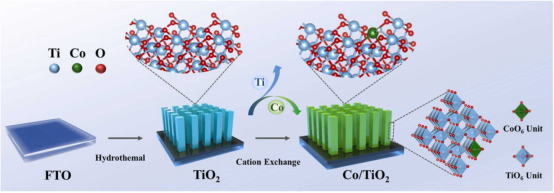
图1. Co/TiO2光阳极的制备示意图。
本 文 要 点
要点一:TiO2光阳极的阳离子置换
Co/TiO2光阳极的制备主要涉及两个步骤,首先通过水热法在FTO衬底上生长垂直排列的TiO2纳米线阵列。然后在TiO2纳米线表面通过气相阳离子置换技术构建Co单原子。具体过程为:将TiO2光阳极置于石英管(直50 mm)中心,将CoCl2粉末置于石英管中心上气流方向约4 cm处。将石英管抽空至负气压后,引入50 sccm N2作为载气。将炉子分别以20 ℃ min-1的升温速率加热到360℃、380℃、400℃和420℃的温度并保持2 min。在相对低的温度和真空惰性气氛下,CoCl2蒸汽同TiO2发生置换发应,控制反应温度可以调节反应速率,进而实现精确的置换控制。如图2所示,XANES结果确认了通过气相阳离子交换的方法在TiO2纳米棒阵列表面构筑的Co为分散孤立的原子态。
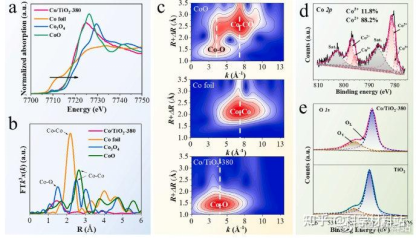
图2.(a)Co K-edge XANES 图谱和(b)Co/TiO2-380 样品和 Co 箔、CoO和Co3O4对比对应的Co K-edge EXAFS 图谱的傅里叶变换图谱,(c)CoO、Co箔和Co/TiO2-380的k3加权EXAFS光谱的小波变换,(d,e)Co/TiO2-380 中Co 2p和TiO2以及Co/TiO2-380 中 O 1s 的高分辨率XPS图谱
要点二:综合光电催化性能测试
光电催化实验证明,样品Co/TiO2-380的光电流密度在1.23VRHE时为1.47 mA cm-2,是TiO2(即0.48 mA cm-2)的约3.1倍。此外,与纯TiO2相比,它的起始电位(VOnset)表现出显著的负偏移,达到0.18 VRHE。这表明原子级分散的Co可以有效地提高水氧化反应动力学,从而导致起始电位的负移。在1.23 VRHE的固定偏压下,Co/TiO2-380光阳极的光电流在100 h内仅下降4.1%,表明其具有出色的稳定性。

图3.(a-b)分别为纯TiO2和样品Co/TiO2-380的J-V曲线和施加的偏置光子电流效率 (ABPE),(c)纯TiO2和样品Co/TiO2-380在1.23 VRHE下的气体产生量和法拉第效率,(d)Co/TiO2-380光阳极的长时间稳定性测试,(e)近年报道的基于TiO2光阳极的性能比较。
要点三:性能提升机制分析
IPCE结果表明,样品Co/TiO2-380在340至400 nm的紫外光区域的IPCE值远高于纯TiO2和Co/TiO2-400。特别地,在340 nm处的最大IPCE高达54%,是纯TiO2的约1.7倍。与纯TiO2相比,Co/TiO2-380光阳极在低偏压区域表现出增强的电荷传输效率,而在高偏压下没有显著差异(1.23 VRHE时均约为94.0%),这是因为Co位点提高在TiO2光阳极的水氧化性能性能,提高低偏压下的电荷传输效率,高偏压时绝大多数的载流子均被用于水分解,无法进一步提升。此外,TiO2和Co/TiO2-380的表面电荷分离效率在1.23 VRHE偏压时分别为47.3%和75.5%,表明原子分散的Co可以增强TiO2光阳极表面的载流子分离。
进一步基于DFT理论计算研究了其水氧化强化机理。对于纯TiO2,其OER的吉布斯自由能分布几乎在三个不同的位置重叠(U = 1.23 V)。这表明纯TiO2表面上的每个Ti位点进行等效计算,用Co代替Ti0位点是来进一步分析具有代表性。此外,TiO2模型的决策步骤(Rate Decision Step,RDS)是*OH到*O的电化学步骤,有1.22 eV的能垒。在用Co代替Ti0位点后,发现Co位点上的整个反应路径与Co/TiO2模型中的Ti2和Ti3位点相比要小得多,表明Co位点处OER反应的能垒降低。根据计算结果,*OH到*O的转换是Co/TiO2水氧化反应的RDS。此外,平坦的ΔG 曲线也表明Co/TiO2上的Co位点更适合实现具有较小水氧化过电位。以上计算结果说明原子分散的Co可以增强TiO2光阳极表面水氧化动力学。

图4.(a)纯TiO2、Co/TiO2-380 和 Co/TiO2-400 在 1.23 VRHE 的 IPCE,(b)标准 AM1.5G 太阳光谱和光阳极的理论吸收光谱,(c-d)纯TiO2 和Co/TiO2-380 的载流子注入效率和载流子分离效率。
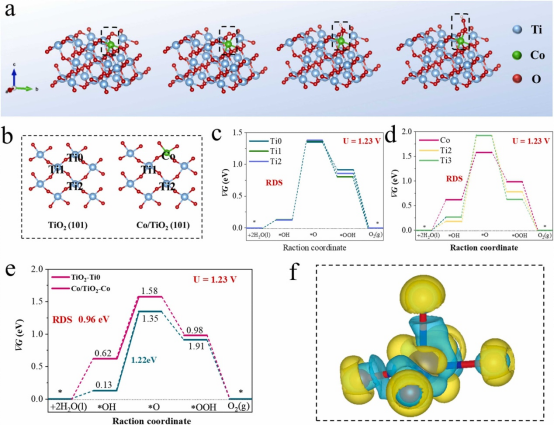
图5.(a)DFT计算的分子结构模型和反应中间产物吸附模型(b)基于纯TiO2和样品Co/TiO2模型的吉布斯自由能计算的吸附位点示意图,(c)纯TiO2的Ti0、Ti1和Ti2位点上水氧化反应中间体的吉布斯自由能图,(d)Co/TiO2样品上 Co、Ti1和Ti2位点上水氧化反应中间体的吉布斯自由能图,(e)水氧化反应中间体在原始TiO2上的TiO2和样品Co/TiO2上的Co位点上的吉布斯自由能图,(f)Co/TiO2分子模型的Co位点的差分电荷图,黄色和蓝色分别表示电子积累和耗尽。
文 章 链 接
https://zhuanlan.zhihu.com/p/619064217?utm_id=0


